第一原理量子化学計算 ab initio calculation
当研究室は、燃料電池をはじめとする電気化学エネルギーデバイスを研究対象としています。スーパーコンピュータによる高度な計算能力を用いることで、その化学反応や材料特性などを第一原理計算と呼ばれる計算手法によって解析しています。以下にその概要を記載しています。
・第一原理計算とは?
ナノスケールでの基本的な原理(=第一原理)である量子力学に基づいて自然現象をコンピュータ上で再現する計算を指します。
・計算の利点と計算に必要なものは?
計算の利点としてはズバリ、高額な実験装置無しで様々な材料物性が(ある程度)予想できる点にあります。下の図に示すように、計算のためには結晶や分子の位置座標・計算機・量子力学の計算コードが必要になります。結晶の位置情報は既にインターネット上で公開されているMaterials projectを代表例としたデータベースからダウンロードすることが可能です。計算機に関しては、計算コードによりますが大学所有のスーパーコンピュータが利用可能です。量子力学の計算コードは代表例として無償版のQuantum espressoもあります。
・計算で何が分かるのか?
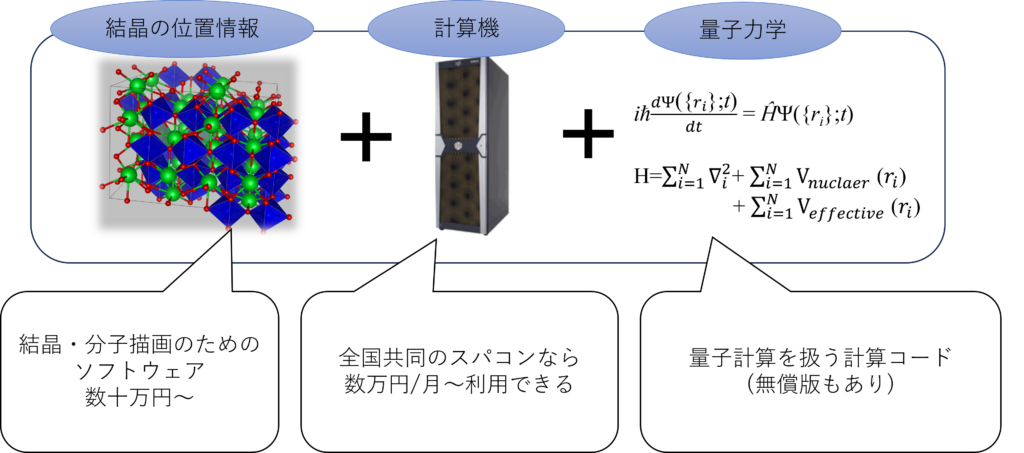
図1、第一原理計算に必要なもの
これまでに様々な材料物性が第一原理計算によって予測されております。その中には実験と良く整合するものあり、下記はそのごく一部になります。
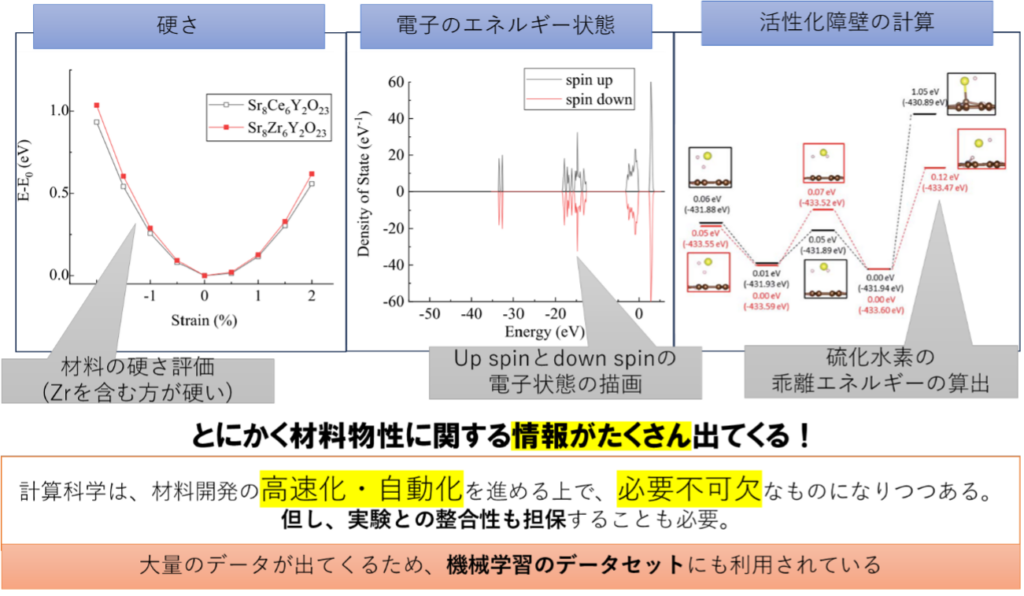
図2、第一原理計算によって分かる事例
左から順に電池材料の硬さの比較、電子状態の解析、また分子の乖離エネルギーの計算事例になります。これらの他にも、様々な物性(電荷、電子マッピング、吸着エネルギーなど)を計算によって評価することも可能です。
第一原理計算に関連する投稿論文: